2022年2月10日,美國 FDA線上舉行了針對信達生物、禮來製藥聯合研發的 PD-1抗體信迪利單抗上市申請的腫瘤藥物諮詢委員會(ODAC)。
出人意料的是,這次專家委員會的最終投票結果為14:1,認為信迪利單抗需要補充額外的臨床試驗,來證明自己在美國的適用性。

來源:ODAC審評文件
最引人注意的,還是 FDA引用了2016年中國食藥監局披露的數據:80%的中國臨床試驗不標準。

來源:ODAC審評文件
信迪利單抗此次上市申請,是國產 PD-1藥物首次出海。但在前期縱享絲滑之後,後期卻迎來 FDA當頭一棒——為什麼出海卡在了審核關?又為什麼投票如此懸殊?值得我們一看。
國產 PD-1的揚帆
2018年12月,由信達生物、禮來製藥聯合研發的信迪利單抗注射液(達伯舒)在中國獲批上市,用於治療復發或難治性經典型霍奇金淋巴瘤(R/R cHL)。
2019年11月,信迪利單抗以降價63%的代價,成為第一個進入國家醫保目錄的 PD-1產品。
受益於進入醫保,信迪利單抗的覆蓋範圍由2019年底的約2000家醫院和300間藥房擴增到截至2020年12月31日的約4000家醫院和900間藥房。得益於此,2020年信迪利單抗的全年銷售額達到22.90億元,較前一年度增加125.4%。
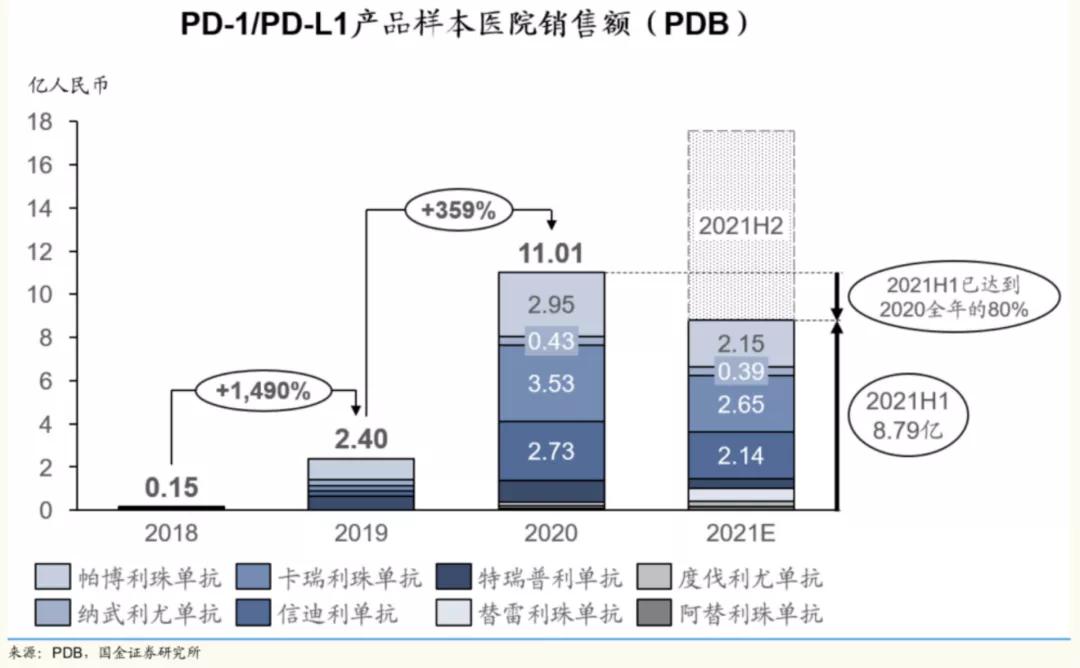
來源:PDB,國金證券研究所
次年3月,信達生物向 FDA遞交了信迪利單抗的上市申請。
2021年5月17日,美國食品藥品監督管理局(FDA)正式受理了信迪利單抗聯合培美曲塞和鉑類用於非鱗狀非小細胞肺癌(NSCLC)一線治療的上市申請。這也是目前中國首個自主研發,上市申請被 FDA受理並進入正式審評階段的生物創新藥。
然而,熱板凳還沒坐上多久,當受理上市申請快進到審核階段,結果卻是壓倒性的「14:1」不通過。
折戟
先來看官方角度,在信達和禮來的這次折戟上,FDA給出了三個解釋。
第一個是臨床格局的變化。
FDA認為,2018年8月20日 Keytruda(帕博利珠單抗)聯合化療即獲批用於治療非鱗狀 NSCLC,並且 K藥臨床數據已經在審批之前廣泛宣傳,但是信迪利單抗的 ORIENT-11實驗在此之後才啟動。
啟動時,一線轉移性肺癌的護理標準已經發生變化,臨床一線療法從化療變為「免疫治療+化療」聯合治療。鑑於 Keytruda(帕博利珠單抗)化療在臨床和統計學上顯示出對總生存期的顯着益處,研究人員不應該將患者納入化療控制組。

來源:ODAC審評文件
第二件事,是 FDA認為單國臨床研究數據不適用於美國患者。
FDA表示,ORIENT-11的研究人群完全由來自一個國家的亞洲患者組成。雖然中國是一個多民族和多民族的國家,但 ORIENT-11研究人群並未反映美國肺癌患者的種族和民族多樣性。
「接受此類研究和類似研究,與行業範圍內對臨床試驗公平代表性的承諾相衝突。」


來源:ODAC審評文件
第三個原因,是它選擇了無進展生存時間(PFS)作為臨床試驗終點,而沒有選擇總生存期終點(OS)。
迄今為止,FDA對轉移性非小細胞肺癌一線免疫治療方案的所有批准都是基於 OS的統計學改善。總生存期被認為是最可靠的癌症終點,並且在可以合理評估時是首選。
所以,FDA表達了對 PFS後繼續用藥的擔憂,表示 ORIENT-11在設計上顯示缺乏多樣性。
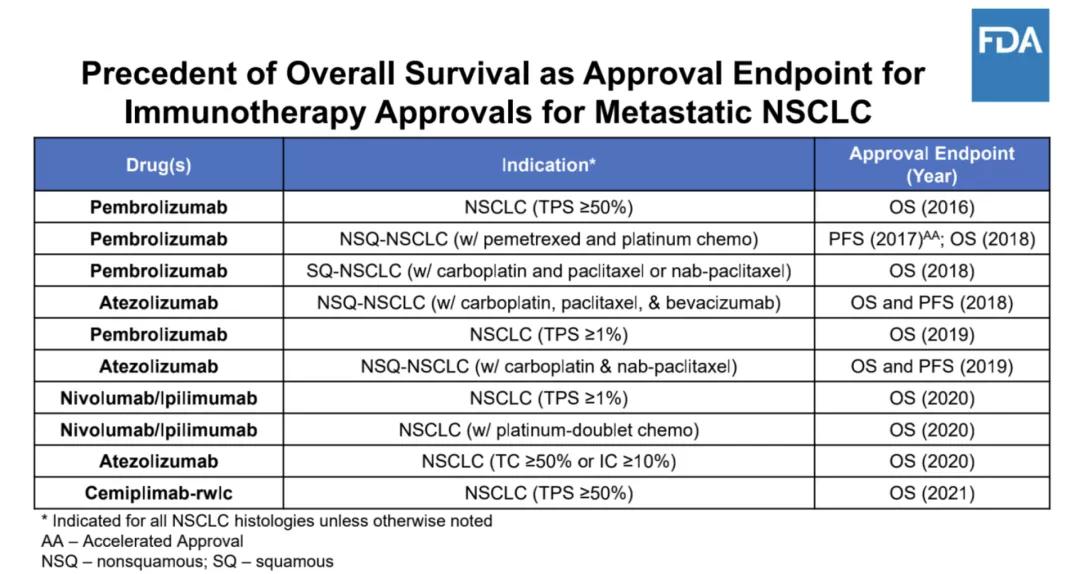
Image來源:ODAC審評文件
另外,FDA還指出,申請人一直沒有就「研究設計或試驗」進行諮詢 FDA。如果諮詢了 FDA,可能會建議將信迪利單抗與 FDA批准的具有總生存期終點(OS)的 PD-1藥物進行頭對頭研究。
風向突變的 FDA
來拉一下時間線。
其實早在2019年,FDA腫瘤學卓越中心主任 Richard Pazdur就曾公開呼籲中國藥企將國產 PD-1/PD-L1抑制劑引入美國市場。
「這對每個人來說都可能是一件好事,因為我們還沒有看到西方製藥公司在價格上有所調整。」他如是說道。
他當時還表示,中國公司模仿 FDA批准產品開發新的 PD-1/PD-L1藥物,獲得 FDA的批准將是沒有太大懸念的,「顯然,它們可能會給出非常相似的結果,所以我們在批准這些藥物時,會很順利。」
在此之後,信達、君實、康方、百濟神州先後向 FDA遞交了上市申請。
然而在2021年,Pazdur卻突然轉變態度。
2021年12月15日,Pazdur在 NEJM上發表文章 The Wild West of Checkpoint Inhibitor Development,指出已上市的 PD-1/PD-L1藥物的適應症中的45%是通過加速審批(Accelerated Approval)這一途徑獲批的,通常是只做了單臂試驗,之後隨機臨床試驗可能顯示出不一致的結果。
「用低於美國臨床對照標準的方式,在中國開發的藥,是不值得 FDA給予照顧來美國上市的。」
在會前一周的2022年2月4日,Pazdur又在柳葉刀上發表題為 Importing oncology trials from China: a bridge over troubled water的文章。
文章指出,至少有25個來自中國的免疫抑制劑類新藥申請,都幾乎只基於在中國做的臨床試驗數據,重申「單一國外數據不能代表美國人口」這一觀點,與2019年那次醫學會議上的公開評論大相逕庭。
不管是疫情初期對中國外科口罩的變臉,還是之前對國產藥物的「無差別抵制」,FDA出爾反爾已經不是第一次。這次的「渣男行為」,對想出海的藥企而言,無疑是當頭一棒,所以就連禮來也吃了一個暗虧。
Fast-Follow創新藥
不過,從藥物本身創新邏輯的角度來看,FDA這一次雖然不厚道,可話糙、理沒那麼糙。因為大部分國內所謂的創新藥,不過是 Fast-Follow的產物。
Fast-Follow名為快速追蹤新藥模式,指在不侵犯他人專利的情況下,在已有靶點和機理的基礎上,對新藥進行分子結構改造或修飾,尋找作用機制相同或相似,具有新治療效果的新藥物。
Fast-Follow包括了 Me-too、Me-better、Me-worse等藥物。
Me-too藥顧名思義,即藥物結構與首創藥相似,只有較小差別,這個較小的差別區分了 Me-too藥和仿製藥。
與仿製藥不同,仿製藥完全照抄原研藥,需要等原研藥專利過期後才能投入市場。但是 Me-too藥由於繞開了專利,即便原研藥仍然在專利期也不需要授權。
Me-better指改良模仿,也就是在原研藥基礎上創新,得到比原研藥更好的療效。
Me-worse是指雖然繞開了原研藥專利,但療效並不如原研藥,現在已經很難獲得 NMPA批准。
如果創新是頭髮,那仿製藥就是鋥光瓦亮的禿頭,Fast Follow藥是三毛——禿了,但沒完全禿。優勢是既能用較少的時間金錢成本開發出新藥,又能規避首創藥的專利保護。
目前,所有的國產 PD-1抑制劑都屬於 Me-too藥。雖然所有的國內廠商都號稱自己的藥是 Me-better藥,即藥物經過改造後,臨床效果要好於首創藥,但可惜臨床試驗的結果並不能自證。
這也正是國內創新藥市場的一個縮影——創新性不夠。
已有的大部分創新藥,都是基於熱門靶點的研究,跟隨首創藥的「小打小鬧」。在中國,同類藥物想要打敗原研藥,可以依靠 Fast-Follow的成本優勢,這其中,想要快速搶佔市場更快的上市至為關鍵。
如果沒有2019年的醫保談判,在「First-in-class」藥廠吃肉之餘,追隨前者腳步的 Me-too藥廠依靠喝湯也能活得十分愜意。但一輪又一輪的醫保談判就像一面「照妖鏡」,藥廠的創新能力在「照妖鏡」面前一目了然。
於是藥企又想出了一招——出海。
這次誰也沒想到的是,反覆無常的 FDA又給藥企們上了一課。
市場競爭背後,患者才是主體
截至去年12月,NMPA已經批准了12款 PD-1/PD-L1產品,涉及11個癌種,44個適應症。其中非小細胞肺癌:MSD、恆瑞、百濟神州、信達均獲批了一線鱗癌和腺癌。
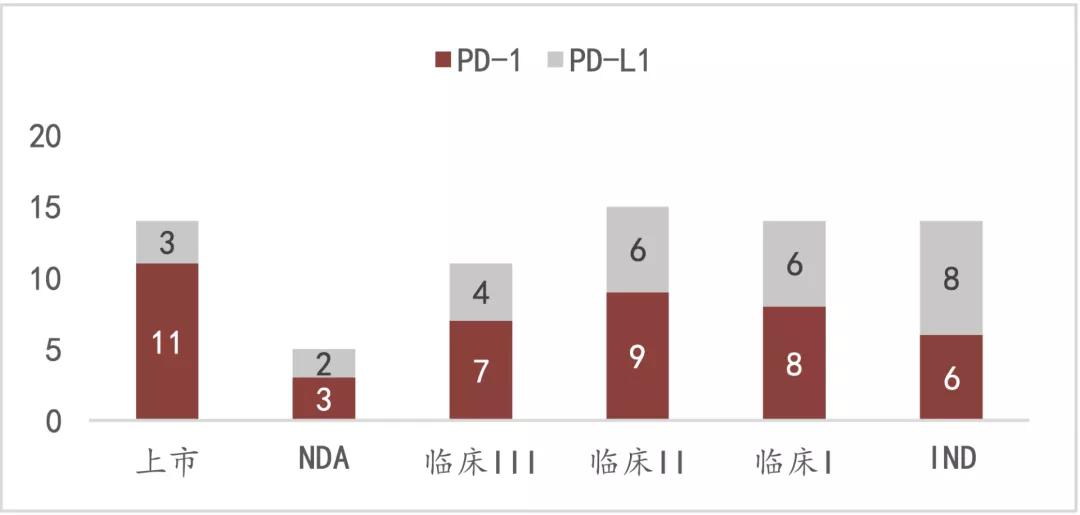
國內 PD-1/PD-L1臨床研究數量來源:FIC intelligence, Insight,莫尼塔研究
PD-1的市場競爭,已經完全進入白熱化階段。
而國內 PD-(L)1抑制劑市場能否規避「內卷」的危害,已成為一個迫切需要回答的問題。
在目前已批准的44個適應證中,有25個來自國產 PD-(L)1抑制劑,其中16個是重複適應證,僅有9個為獨家或同類首個獲批適應證。
與之形成鮮明反差的是,在4個進口 PD-(L)1抑制劑獲批的19個適應證中,有18個仍是獨家或是同類首個獲批適應證。
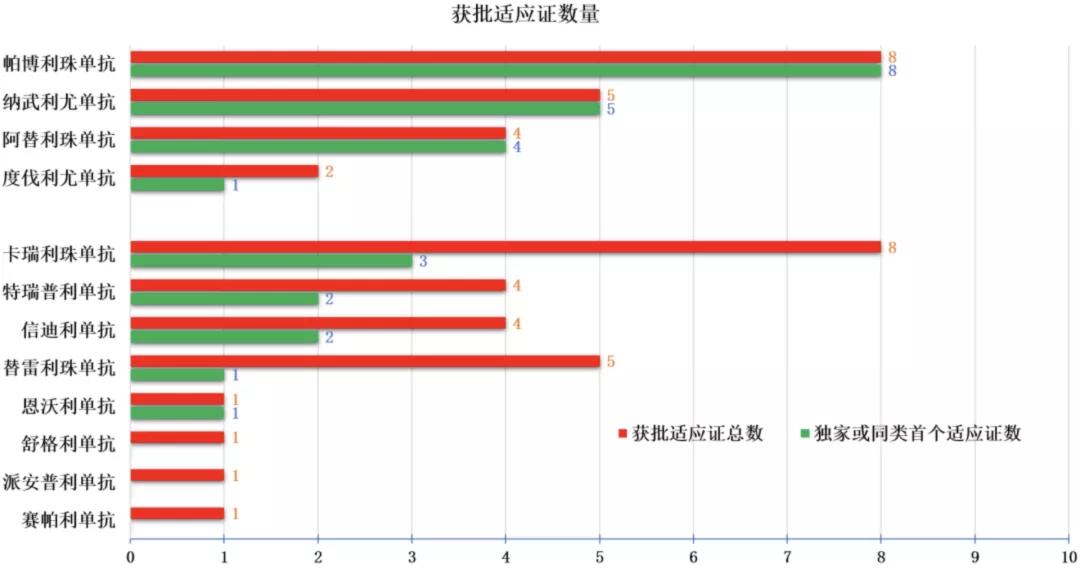
PD-(L)1抑制劑國內獲批適應證數量及獨家或首個適應證數量,「統計標準以獲批受理號為準」,圖片來源:文獻整理
那些本就創新不足的國產 PD-(L)1抑制劑,所開展的新臨床研究,能否滿足患者之前未被滿足的需求,將會是接下來藥企們最需要交的答卷。







{kind=link}